
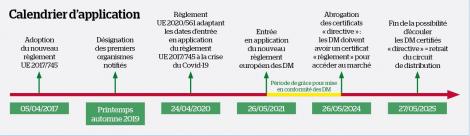
Les dispositifs médicaux – cette grande famille hétérogène de produits de santé qui inclut aussi bien les lunettes de vue que les fauteuils roulants, les préservatifs, les couronnes dentaires, les appareils d’échographie ou les compresses – vont connaître une évolution sans précédent avec l’entrée en application du nouveau règlement européen qui leur est consacré. Exit les anciennes directives européennes prises depuis les années 1990, la refonte est totale. Un travail mené depuis 2012 a abouti à l’adoption du règlement européen en 2017 et à une mise en application initialement prévue le 26 mai 2020, reportée d’un an en raison de l’épidémie de Covid-19. Une année de report où l’on aura rarement autant parlé de dispositifs médicaux : masques, respirateurs, tests antigéniques…
Les changements introduits par ce nouveau règlement européen sont multiples. La classification – classes I, IIa, IIb et III – en fonction du niveau de risque lié à l’utilisation du dispositif médical (DM) a fait l’objet d’un grand nettoyage : les règles ont été révisées et de nouvelles sont apparues, conduisant au passage de certains DM dans une classe plus élevée et à l’arrivée de dispositifs jusqu’alors catégorisés comme non médicaux telles que les lentilles de contact non correctrices ou les produits de comblement des rides. Ces modifications ont contraint les fabricants à passer en revue leurs gammes de produits pour vérifier leur classe d’appartenance et… faire des choix.
Une classification plus élevée entraîne en effet davantage d’exigences. Ainsi, un DM de classe I, dont la vérification de conformité incombe au seul fabricant, qui passe dans une classe supérieure doit être contrôlé par un organisme notifié (ON). Et là encore, la liste des exigences a été allongée et renforcée, autant pour les ON chargés d’attribuer le marquage CE médical, que pour les opérateurs (fabricants, mandataires et distributeurs) et donc pour les dispositifs médicaux qui doivent être évalués par les uns et contrôlés par les autres. Même ceux qui ne changent pas de classe vont désormais devoir répondre aux exigences du règlement européen. « Le niveau d’exigences augmente pour tous les acteurs. Pour les fabricants, l’évaluation clinique est renforcée, tout comme les contrôles liés à la certification », explique Cécile Vaugelade, directrice des affaires réglementaires du Syndicat national de l’industrie des technologies médicales (SNITEM).
Statut de distributeur
Et les pharmaciens ? Ils héritent de nouvelles obligations au titre de leur statut de distributeur, réunies dans l’article 14 du règlement européen. Avant la mise à disposition d’un DM, ils doivent vérifier qu’il porte le marquage CE et qu’il a sa déclaration de conformité européenne, que les informations obligatoires pour le fabricant (étiquetage, notice), les coordonnées de l’importateur et du mandataire s’il s’agit d’un DM fabriqué hors de l’Union européenne, et l’identifiant unique (IUD) sont bien présents. Ils doivent également s’assurer de respecter les conditions de transport et de stockage déterminées par le fabricant. « Avant le règlement européen, il était déjà sous-entendu que chacun doit distribuer des produits conformes. Ce qui change c’est que le texte précise les vérifications à effectuer et comment les faire », ajoute Cécile Vaugelade. Pour Sophie Valentin, pharmacien qualité chez Oxypharm, les pharmacies habituées aux procédures qualité n’auront pas de difficulté. Malgré tout, plusieurs pistes sont à l’étude pour mettre en place un système d’accompagnement pour les officines qui en feraient la demande à Oxypharm.
Des précisions utiles étant donné la diversité des distributeurs de dispositifs médicaux, mais qui ne devraient pas poser problème aux pharmaciens. « Ce sont des professionnels de santé qui dispensent les médicaments, ces obligations concernant les DM entraient donc déjà dans leur cœur de métier. C’est du simple bon sens, l’article 14 est très descriptif dans les vérifications permettant de s’assurer a priori qu’un DM est conforme, il faut le prendre comme un guide », explique Thierry Sirdey, à la tête de la direction des dispositifs médicaux à l’Agence nationale de sécurité du médicament et des produits de santé (ANSM). Attention, prévient néanmoins Me Muriel Artis, avocate spécialisée en droit de la propriété intellectuelle, « en cas de non-respect de l’une de ces conditions, le distributeur doit refuser de mettre le dispositif sur le marché ». Cela nécessite « la mise en œuvre de procédures de vérification » qui doivent être traçables, à réaliser « par échantillonnage et en sollicitant contractuellement de l’opérateur précédent les informations et documents permettant de procéder à la vérification ». Aussi, souligne Me Artis, « le distributeur devient le surveillant du fabricant qu’il devra informer s’il estime que le DM n’est pas conforme, et même un dénonciateur en cas de risque grave ou de dispositif falsifié du fait de l’obligation d’en informer l’autorité compétente », en l’occurrence l’ANSM.
Rationalisation de gammes
Le nouveau règlement prévoit aussi une obligation de déclarer les incidents de matériovigilance pour les distributeurs, une pratique déjà bien connue du pharmacien. Le spécialiste du matériel médical Oxypharm, également classé dans les distributeurs dans la réglementation européenne en tant que prestataire, a mis en place une procédure de signalement depuis de nombreuses années. « Nous travaillons en partenariat avec le pharmacien qui nous remonte les informations et que nous remontons nous-mêmes jusqu’au fabricant », précise Sophie Valentin. « La coopération entre les acteurs économiques sur les réclamations, les signalements et la traçabilité est essentielle. Les pharmaciens y sont déjà habitués avec les médicaments. »
Ces changements sont lourds à mettre en œuvre pour les fabricants qui sont, selon le dernier panorama réalisé par le SNITEM en 2019, pour 94 % d’entre eux des petites ou des très petites entreprises. « Ils ont dû réaliser un gros travail de screening sur leur portefeuille pour connaître leurs priorités d’action, pour se concentrer d’abord sur les produits dont le certificat arrive à échéance à court terme, évaluer ceux qui vont changer de classe, puis sur la révision complète de toutes leurs documentations techniques. Et effectivement, il y a des rationalisations de gamme pour certains produits en fin de vie car les efforts à fournir ne valent pas le coup », détaille Cécile Vaugelade, qui rappelle que le SNITEM essaie d’accompagner le plus possible l’ensemble des entreprises de DM.
Période de grâce
Conséquence à l’officine : certains DM peuvent disparaître par choix d’un arrêt de commercialisation à la date de fin de leur certificat « directive » et au plus tard le 26 mai 2024. Une période de grâce a en effet été mise en place, non seulement pour laisser du temps aux fabricants, mais aussi pour permettre aux ON, nouvellement désignés et encore peu nombreux, d’absorber toutes les certifications « règlement » à réaliser. Ainsi, seuls les DM de classe I doivent disposer d’un certificat « règlement » au 26 mai 2021. Les autres classes peuvent bénéficier de leur ancien certificat « directive » tant qu’il est valide et au plus tard jusqu’au 26 mai 2024. À cette date, tous les certificats « directive » seront abrogés et les produits n’ayant pas obtenu un certificat « règlement » ne pourront être mis sur le marché. Cependant, le règlement européen différencie les obligations du fabricant et du distributeur : les DM certifiés « directive » et non « règlement » déjà dans le circuit de distribution au 26 mai 2024 pourront continuer à être écoulés jusqu’au 27 mai 2025. Il n’y aura donc pas de retrait du marché en officine avant cette date.
Cette entrée en application progressive du règlement va entraîner la cohabitation en pharmacie de DM pourtant similaires mais de classe différente car certains dépendront encore de l’ancienne directive tandis que d’autres seront certifiés « règlement ». « Les pharmaciens doivent accepter cette cohabitation, sans quoi nous aurions des problèmes d’approvisionnement. L’important pour eux est de s’assurer que le produit est conforme et a le bon certificat. En cas d’incertitude, les officinaux peuvent se référer au guide publié sur le sujet et coécrit avec l'association des pharmaciens hospitaliers Euro-Pharmat, sous la forme de questions-réponses », précise Cécile Vaugelade.
Éviter l’embouteillage de 2024
Les pharmaciens pourront aussi être confrontés à des ruptures de stock pour des DM dont le certificat « directive » est arrivé à échéance mais dont le certificat « règlement » n’a pas encore été délivré. Avec seulement une vingtaine d’ON désignés en Europe (procédure en cours pour 40 autres ON), les délais d’évaluation peuvent s’allonger avant d’obtenir le fameux sésame. D’ailleurs, prévient le directeur des DM de l’ANSM, il faut à tout prix éviter un embouteillage en 2024, lorsque l’ensemble des certificats « directive » seront abrogés. « Même si le certificat " directive " est encore valide, l’anticipation est le maître-mot. Les fabricants doivent déjà travailler sur la mise à jour de la documentation technique et sur les nouvelles exigences d’évaluation clinique. » D’une part, le SNITEM et l’ANSM finalisent une procédure de gestion de toutes les ruptures (et pas seulement celles liées au certificat), retardée par l’épidémie de Covid-19, qui pourrait se mettre en place au second semestre 2021. D’autre part, l’ANSM développe une procédure de gestion des certificats. « Ce n’est pas parce qu’un certificat est arrivé à échéance que le produit devient dangereux le lendemain. Nous avons des solutions, notamment avec un dispositif de prorogation de certificats dans certains cas précis lorsque l’ON nous garantit que l’évaluation est en cours », indique Thierry Sirdey.
Les pharmaciens pourront se référer à la base de données EUDAMED pour obtenir toutes les informations qu’ils recherchent sur un dispositif médical. Celle-ci, administrée par la Commission européenne, devrait être pleinement opérationnelle en mai 2022.
A la Une
Gel des prix sur le paracétamol pendant 2 ans : pourquoi, pour qui ?
Salon des maires
Trois axes d’action pour lutter contre les violences à l’officine
Médication familiale
Baisses des prescriptions : le conseil du pharmacien prend le relais
Caisse d’assurance vieillesse des pharmaciens
Retraite des pharmaciens : des réformes douloureuses mais nécessaires